Von Struktur-Eigenschafts-Beziehungen zu Prozess-Struktur-Eigenschafts-Beziehungen in weicher kondensierter Materie – ein computerbasierter physikalischer Ansatz
Forschungsbericht (importiert) 2014 - Max-Planck-Institut für Polymerforschung
Von der Biologie bis zur Fotovoltaik ist die Entstehung von makromolekularen Strukturen und deren Funktionalität das Ergebnis von Nichtgleichgewichtsprozessen. Obwohl von zentraler Bedeutung, fehlt ein grundlegendes molekulares Verständnis dieser Prozesse. Die Entwicklung der letzten Jahre ermöglicht es nun, solche Prozesse über viele Zeit- und Längenskalen zu verfolgen. MolProComp greift diese Entwicklung auf und wird Computersimulationsmethoden entwickeln, die auf die Analyse, die Kontrolle und die Manipulation solcher Prozesse abzielen.
Die Entstehung makromolekularer Materialien ist immer das Ergebnis eines Relaxationsprozesses in einen (meta-) stabilen Zustand. Da dieser Zustand in den seltensten Fällen dem thermodynamischen Gleichgewicht entspricht, ist er vom Weg im Zustandsraum, d. h. von der Prozessführung mit angelegten Temperaturen, Verzerrungen usw. abhängig. Nur der sogenannte Gleichgewichtszustand ist unabhängig von der Prozessführung. Das gilt für alle Vorgänge in der Biologie, aber auch in ähnlicher Form für alle Materialien/Systeme des täglichen Gebrauchs, von Lebensmitteln bis zu Plastik oder Stahl. Ein klassisches Beispiel für die Langlebigkeit solcher metastabilen Zustände sind Gläser. Hier haben die Scheiben mittelalterlicher Kathedralen oft 1000 Jahre überstanden, ohne ihre Schönheit eingebüßt zu haben. Weitere Beispiele sind Stähle, wie z. B. die in Abbildung 1 gezeigte Damaszenerklinge, die durch aufwändiges Schmieden entstanden ist, aber in dieser Form nie durch einfaches Abkühlen eines glühenden Werkstücks entstehen würde.
Der Menschheit ist die Bedeutung der Prozessführung, also des Herstellungsverfahrens, seit jeher bewusst. Da aber meist von der makroskopischen Struktur ausgegangen wird, ist der Anteil empirischer Verfahren sehr hoch. Auch bei langkettigen Polymeren stellen sich viele Fragen dieser Art. Nimmt man z. B. ein Polymer, das bei niedrigen Temperaturen kristallisiert (z. B. Polyethylen), dann hängt die Größe der entstehenden Kristallite nicht nur von der Temperatur ab, sondern auch ganz entscheidend von der Konzentration der Polymere während der Kristallisation. In der Schmelze bleiben die Kristallite recht dünne Plättchen, während aus verdünnter Lösung entstandene Kristalle viel größer werden können. Die Ursache liegt in der unterschiedlichen Zahl von Kettenverknotungen, die in der Schmelze hoch und in der Lösung niedrig sind und die die Kristallisation behindern. Eindrucksvoll demonstriert wurde das schon vor einiger Zeit an Polymerketten mittlerer Länge von Spiess und Rastogi [1]. Wie man solche Effekte (im Prinzip) systematisch einsetzen kann, beweist uns die Biologie.
, Perlmutt (Mitte; nach MIT Techn. Rev. 2008) sowie die unterschiedliche Organisation von Hexabenzocoronenen je nach Seitengruppen und Lösungsmitteln (rechts; K. Müllen, MPI für Polymerforschung).")

Abbildung 1 (Mitte) zeigt die Struktur von Perlmutt, einem sehr widerstandsfähigen Material aus Proteinen und anorganischen Kristalliten. Die Eigenschaften solcher Kompositmaterialien hängen von den Details der Kristallitpackung, den Eigenschaften der organischen Zwischenschichten sowie der Wechselwirkung (Verklebung) der Polymere mit den Kristalliten ab. Neben der Frage nach den Eigenschaften ist es von besonderem Interesse herauszufinden, wie die Aggregation der unterschiedlichen Komponenten und unter Umständen das anisotrope Wachstum einzelner Kristallite auf molekularer Ebene verstanden, kontrolliert und gesteuert werden kann. Genau hier setzt das Projekt „MolProComp“ an, das solche Vorgänge mittels Computersimulationen untersuchen wird.
Warum „Weiche Materie“?
Polymere, Biomoleküle, molekulare Aggregate und kolloidale Systeme werden heute oft unter dem Oberbegriff „Weiche Materie“ zusammengefasst. Der Begriff veranschaulicht einen zentralen Unterschied zu konventionellen anorganischen Materialien, die Systeme sind „weich“. Das macht sie für unser Vorhaben besonders geeignet. Hinzu kommt ihre Bedeutung in allen Bereichen unseres Lebens. „Harte Materie“ zeichnet sich unter anderem dadurch aus, dass die charakteristische Längenskala, die z. B. eine Kristallstruktur beschreibt, in der Größenordnung eines Atomdurchmessers liegt. Da jedes Atom mit mehreren Nachbarn wechselwirkt, erreicht man eine sehr hohe Energiedichte – ein direktes Maß für die Größenordnung der elastischen Konstanten. Nimmt man dagegen eine Polymerschmelze oder eine Biomembran, so kann man das einzelne Monomer, also die Wiederholungseinheit, aus denen die Polymere aufgebaut sind, oder das einzelne Lipid als „Pseudoatom“ im Rahmen dieser Argumentation betrachten.
![Abb. 2: Links ist die Relaxation aus einem kristallinen Zustand in eine amorphe Schmelze mit und ohne den Einfluss der Verhakungen gezeigt [1]. Rechts](https://www.mpg.de/11613087/original-1508157539.jpg?t=eyJ3aWR0aCI6ODQ4LCJmaWxlX2V4dGVuc2lvbiI6ImpwZyIsIm9ial9pZCI6MTE2MTMwODd9--cc13cde55bb45773ffc461e3d4459e1ed810b98b "Abb. 2: Links ist die Relaxation aus einem kristallinen Zustand in eine amorphe Schmelze mit und ohne den Einfluss der Verhakungen gezeigt [1]. Rechts ist die langsame Relaxation kollabierter sehr langer Ketten durch langsames Verhaken mit der (nicht gezeigten) Umgebung als Ergebnis einer Simulation dargestellt [3].")
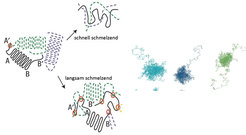
Abb. 2: Links ist die Relaxation aus einem kristallinen Zustand in eine amorphe Schmelze mit und ohne den Einfluss der Verhakungen gezeigt [1]. Rechts ist die langsame Relaxation kollabierter sehr langer Ketten durch langsames Verhaken mit der (nicht gezeigten) Umgebung als Ergebnis einer Simulation dargestellt [3].
Neben dem Größenunterschied zu einzelnen Atomen (etwa Faktor 10 und mehr) und der typischerweise niedrigeren Zahl von Nachbarn ergibt sich eine um mindestens den Faktor 1000 geringere Energiedichte. Das heißt, solche Systeme sind mindestens 1000fach „weicher“ als konventionelle Festkörper [2]. Genau das hat entscheidende Vorteile, da die relevante Längenskala von einigen Nanometern für moderne Experimente (z. B. AFM: Atomic Force Microscopy) direkt erreichbar ist. Andererseits macht die „Weichheit“ die Systeme auf molekularer Ebene langsamer, was eine experimentelle Analyse und Manipulation erleichtert. Parallel zu den experimentellen Fortschritten der letzten Zeit sind skalenübergreifende Computersimulationsmethoden entwickelt worden, die es erlauben, Strukturbildungsprozesse auf molekularer Ebene zu verfolgen und in nie gekannter Präzision zu analysieren. Zu diesen Entwicklungen hat die Theoriegruppe des MPI für Polymerforschung entscheidend beigetragen, so dass wir jetzt in der Lage sind, diese Herausforderung anzunehmen. Dabei geht es um eine neue Sichtweise der Materialentstehung, bei der die Entstehungsprozesse und deren Kontrolle und Manipulation auf molekularer Ebene im Vordergrund stehen (Abb. 2).
Im Rahmen des Projektes MolProComp werden drei Schwerpunkte gesetzt:
- Strukturbildung und Rheologie in Polymerschmelzen extrem langer Ketten
- Simulationen zur gesteuerten Strukturbildung in organisch-anorganischen Kompositmaterialien (Biomineralisation)
- Strukturbildung in mehrkomponentigen (Schicht-)Strukturen in der organischen Elektronik
Polymerschmelzen
Wie schon vorher erwähnt, hängen die rheologischen Eigenschaften von Polymerschmelzen sehr vom Verhakungszustand der Ketten ab. Ein Weg, diesen im Ausgangsmaterial zu ändern, ist die Verwendung von dichten Polymeren, die aus separierten Kristalliten einzelner Ketten bestehen. Ein solcher Weg ist allerdings sehr aufwändig und auf kristallisierende Polymere beschränkt. Alternativ kann man individuell kollabierte Polymerknäuel zu einer Schmelze verdichten. Bei kurzen Ketten ergibt das keinen deutlichen Vorteil, da die Ketten sehr schnell ins Gleichgewicht relaxieren (äquilibrieren) und die Struktur verknäuelter und verknoteter Fäden annehmen. Für sehr lange Ketten, etwa oberhalb dem 50fachen des charakteristischen Verhakungsmolgewicht (also etwa M > 70.000 (PE-Polyethylen), 900.000 (PS-Polystyrol)) ist dieser Prozess sehr stark verzögert [3]. Das Verhakungsmolgewicht gibt an, ab welcher Kettenlänge Verknäuelungen der Ketten für die rheologischen Eigenschaften dominieren. In der Praxis würde das bedeuten, dass Schmelzen kollabierter Knäuel relativ einfach technisch zu verarbeiten sind, während das bei äquilibrierten Schmelzen sehr schwierig und energieintensiv ist. Einer Frage, der wir im Rahmen von MolProComp nachgehen, ist, wie man den Prozess der Relaxation ins Konformationsgleichgewicht steuern kann, um so zu optimal verarbeitbaren Werkstoffen aus extrem langen Polymeren mit optimalen Endeigenschaften zu kommen.
Organisch-anorganische Kompositmaterialien
Während man im ersten Fall theoretische Konzepte nutzen kann, die es zulassen, vereinfachte Polymermodelle zu untersuchen, ist das im Falle der Biomineralisation nur sehr eingeschränkt möglich. Neben dem Aspekt der Morphologie auf mesoskopischer Ebene müssen schon die Strukturbildungspfade systematisch mit zum Teil atomistischer Auflösung untersucht werden. Das verlangt neben generischen Modellen Studien unter Einbeziehung von chemischen Details auf unterschiedlichen Ebenen. Besonders wichtig werden Simulationen mit variabler Auflösung im Raum. An Stellen, an denen Aggregation und/oder strukturbildende Anlagerung von Molekülen stattfindet, ist oft die Berücksichtigung aller chemischen Details notwendig, während anderenorts stark vereinfachte Modelle ausreichen. Mit der in den letzten Jahren in Mainz entwickelten AdResS (Adaptive Resolution Scheme)-Methode [4] ist genau das möglich. Sie erlaubt es, so erstmals völlig offene Systeme mit Molekulardynamik zu untersuchen.
Strukturbildung und organische Elektronik

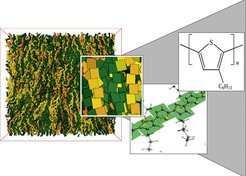
Während in den beiden ersten Themenfeldern mechanische Eigenschaften im Vordergrund standen, sind es hier die elektronischen Eigenschaften polymerer Halbleiter. Dazu ist nicht nur die globale Konformation der Ketten, sondern auch die relative Lage der konjugierten Bereiche auf der Kette von entscheidender Bedeutung. Hinzu kommt die Komplikation, dass Halbleiterpolymere sehr kettensteif und deshalb nicht löslich sind. Dazu müssen flexible Seitenketten als Löslichkeitsvermittler zugefügt werden. Das stellt eine enorme Herausforderung für die Herstellung ganz allgemein dar. Grundlegende Fragestellungen, wie man lokale Packung und globale Morphologie kontrolliert einstellen kann, sind noch nicht gelöst [5]. Bei MolProComp können wir auf den ersten beiden Themenfeldern aufbauen und diese um die Fragestellung der elektronischen Eigenschaften erweitern. Erste Resultate, wie Abbildung 3 zeigt, sind sehr vielversprechend und zeigen, wie die systematische Verbindung von skalenübergreifenden Computersimulationen zur Strukturbildung mit quantenchemischen Rechnungen zu einem molekularen Verständnis der Eigenschaften führen.
Mit der Einbeziehung der Strukturbildungspfade in die Simulation von Materialeigenschaften erwarten wir einen wesentlich besseren Zugang als mit dem bisherigen ausschließlichen Fokus auf die Endeigenschaften. Zumindest für diese Fragestellungen gilt also auch: „Der Weg ist das Ziel“.
Literaturhinweise
Nature Materials 4, 635-641 (2005)
DOI: 10.1038/nmat1437
Faraday Discussions 144, 9-24 (2010)
DOI: 10.1039/B919800H
Macromolecular Theory and Simulations 19, 44-56 (2010)
DOI: 10.1002/mats.200900065
DOI: 10.3390/e16084199
Macromolecules 46, 5762-5774 (2013)
DOI: 10.1021/ma400646a